PLEASE VISIT OUR
COMPUTATIONAL SYSTEMS BIOLOGY (COSBI) GROUP PAGE
We have been working in the field of computational biology and bioinformatics on protein-protein interactions and protein dynamics; specifically on:
SERVERS & DATABASES
Please follow the link to reach our Servers and Databases
Protein-protein interfaces
These are the regions where two proteins bind each other. We have extensively studied the structural (geometrical) and physico-chemical properties of interfaces. We have two interface datasets (Keskin et al, 2004 and Tuncbag et al 2009, PRINT). In these datasets, the protein interfaces are classified as Type I, II and III. She showed that as expected if two proteins are similar, they bind through similar interfaces (type I). She also emphasized that, unexpectedly, even if two proteins are dissimilar, they might still use similar type of interfaces (type II). Still, unpredictably she showed that a single protein can bind to many proteins using a single region on its surface (type III). In the upcoming studies, she extended the folding paradigm to interfaces: Based on the recognition that binding and folding are similar processes with similar underlying principles, she proposed that interface structural similarity exists not only between homologous protein pairs; different protein fold-pairs can also interact via similar interface architectures and these architectures are similar to those observed in single chain proteins. (Publications are highlighted green in this category.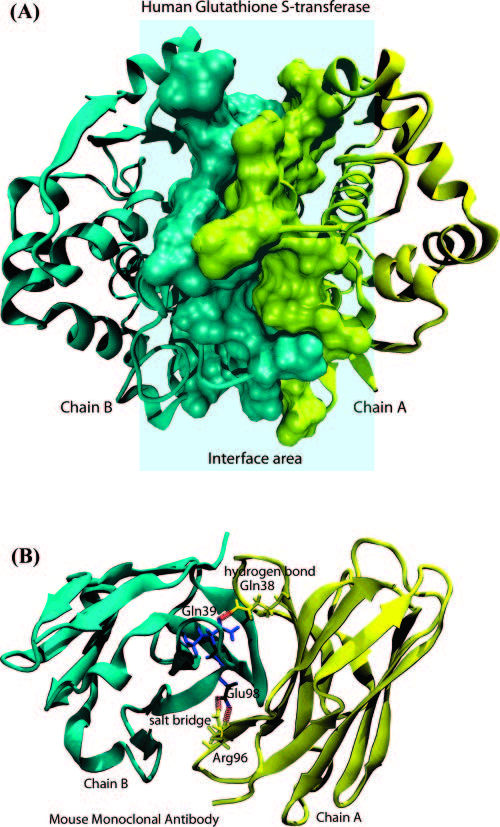
Hot spots at protein interfaces
When two proteins bind each other, individual residues at the interface take place in the interaction. Experimentally, it was shown that some residues contribute more to the binding, whereas some others are not so important. The important residues are called hot spots.We showed that these hot spots are not randomly distributed at the interfaces, but rather form a dense, tight packing region. We called these regions as hot regions. Hot regions are extremely important to understand how proteins recognize each other and they are essential in drug discovery since protein surfaces are large there are infinitely many possible sites that dugs can bind, but in reality this is not the case, and drugs selectively bind these hot regions.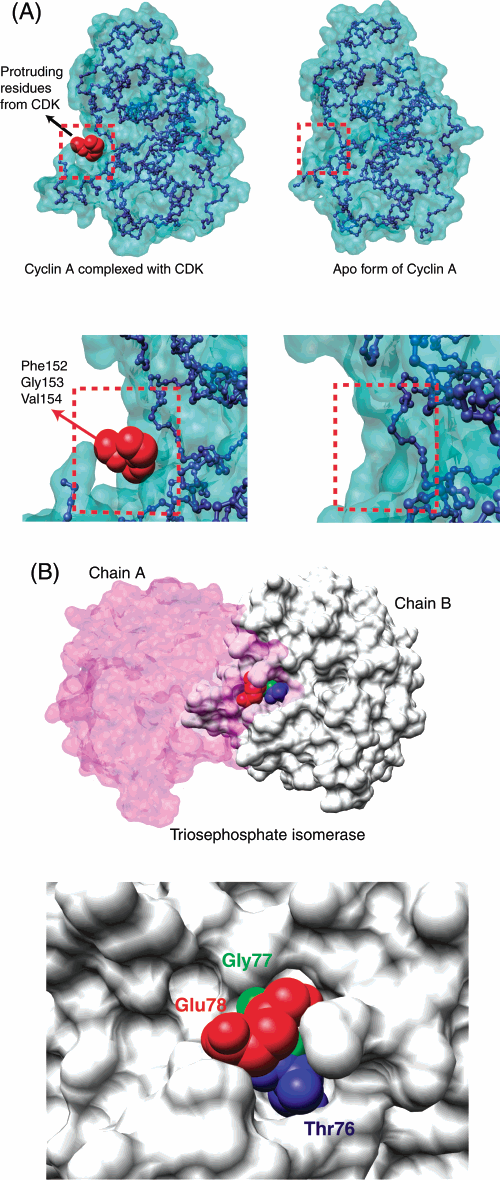
Protein-protein interactions
We developed an algorithm to predict protein-protein interactions inspired by the idea that known 3D interface architectures can be used as templates to identify interacting protein pairs independent of homology or global fold similarity; that is, if two complementary partners of a known protein interface are structurally similar to the surfaces of two unbound proteins, these two proteins may interact. The algorithm is called PRISM and is available at http://prism.ccbb.ku.edu.tr/prism/.
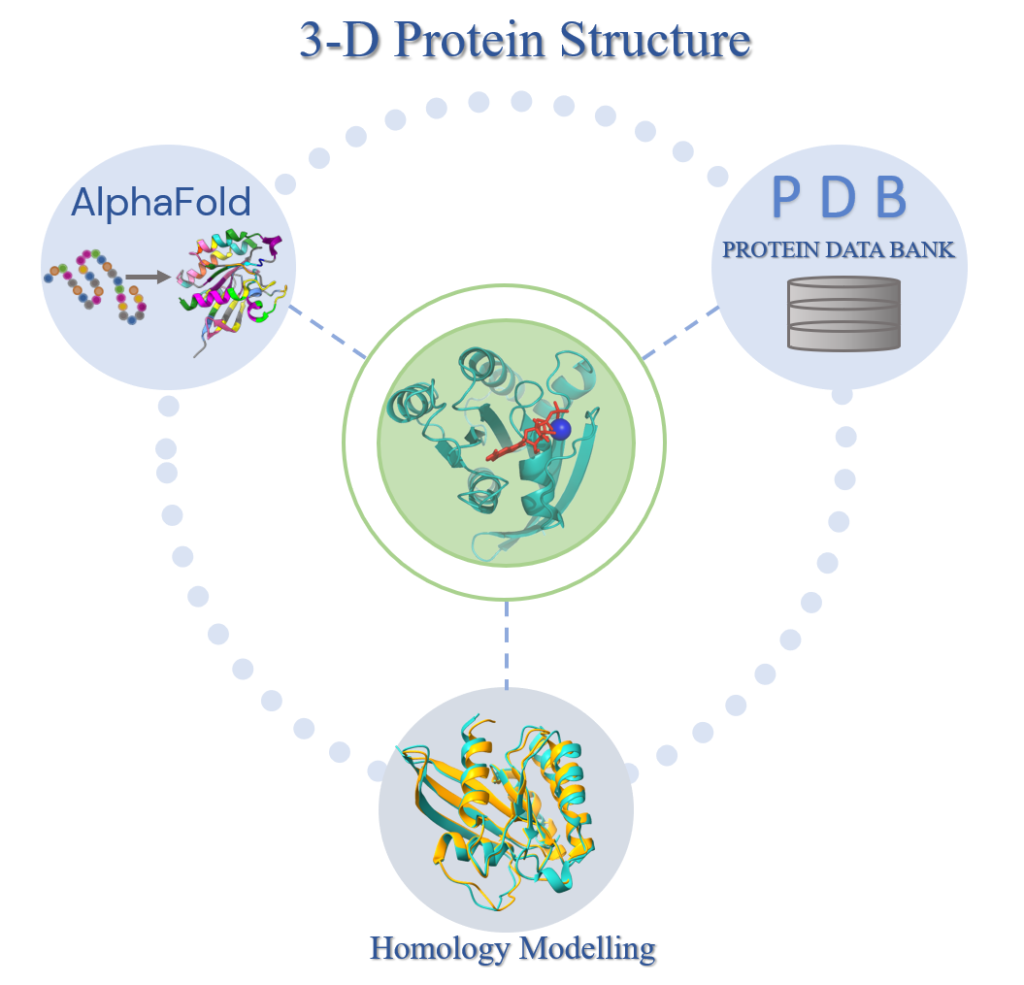
Human Interactome
In this project, we aim to build the structural human interactome via combining available experimental and computational datasets.
The main aim of this project is to extract the comprehensive human interactome, integrate it with the knowledge of 3-dimensional structure (experimental or homology model), make the interactions more physical and meaningful, and present the most comprehensive and reliable structural interactome resource in the literature to the scientific community.
We will apply the PRISM algorithm to each potential interaction to build the cell type, cell cycle, and subcellular localization specific human interactome.
Protein Dynamics
We use normal mode analysis and molecular dynamics simulations to understand the relations between stucture and molecular function. We mostly work on Ras-related pathway proteins.
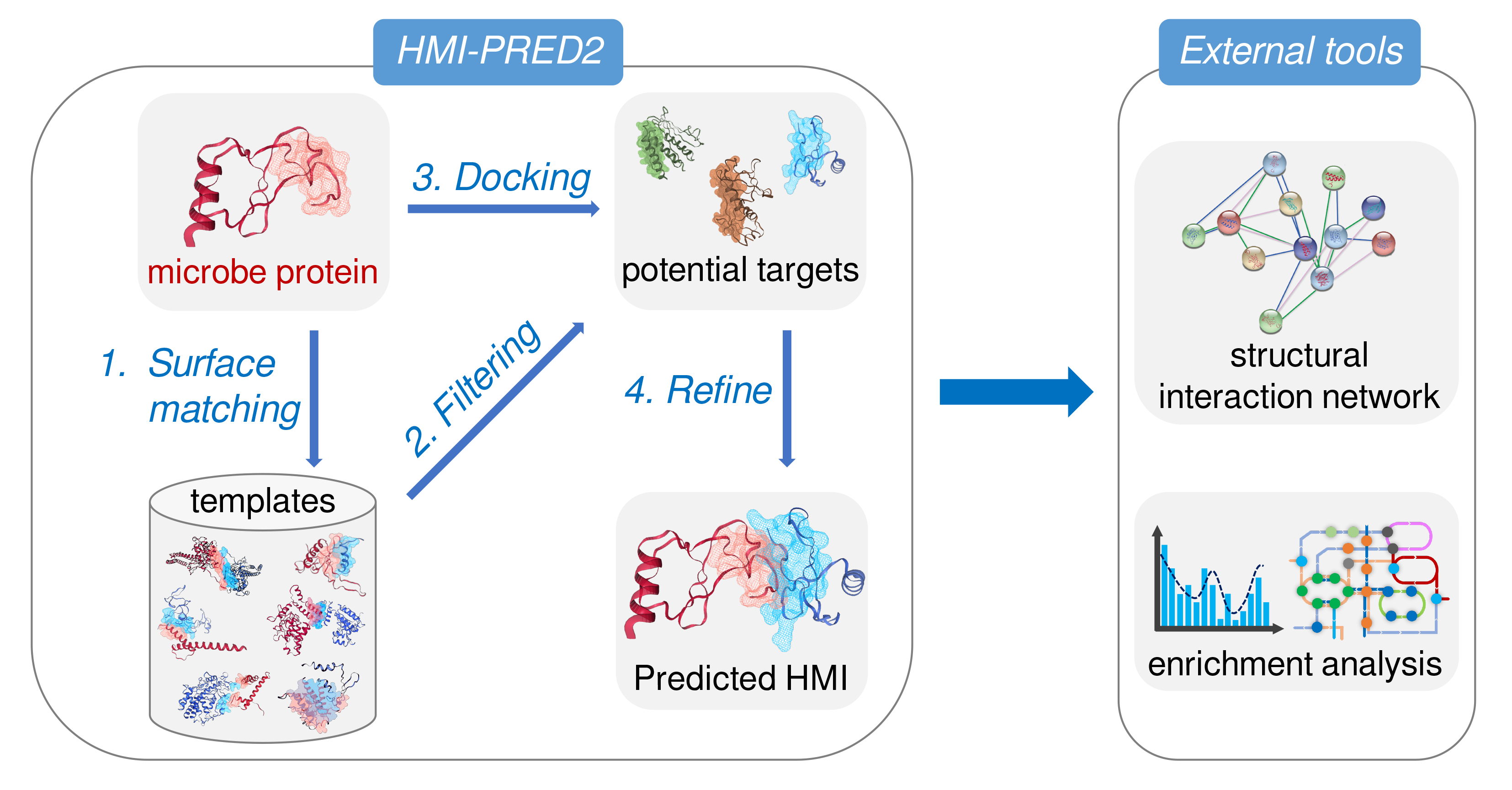
Host-micribiome interactions
HMI-PRED 2.0 predicts microbial target proteins based on interface mimicry. Assume two proteins with a similar surface patch (interface). One of them is known to interact with a target protein by attaching the surface patch to the target. Then, it is possible that the other protein having a similar patch also interacts with the same target. This is interface mimicry, the core concept behind HMI-PRED 2.0. Interface mimicry is an efficient strategy: it allows microbes to affect hosts without strong sequence homology or global structural similarity.